Felix Ngassa Research Interests
Introduction
Studying the influence of adducts stereochemistry on local DNA structure and protein recognition is critical for an increased understanding of polycyclic aromatic hydrocarbon (PAH) mediated carcinogenesis. A major problem inhibiting this study has been the tedious and often cumbersome synthetic route to PAH-nucleoside adducts. With the rapidly growing technique of transition metal catalysis for C-C and C-N bond formation in modified nucleosides, the synthesis of PAH-nucleoside adducts can be attainable in a reasonable manner. The viability of transition-metal-catalyzed reactions demands the investigation of several reaction conditions. These conditions when properly investigated will lead to a generalization of some determining factors for a successful coupling of PAH and modified nucleosides.
Our group focuses on exploring the effect of the catalyst, ligand, base and solvent on the outcome of the coupling reaction of modified nucleosides with aliphatic and aromatic substrates, as well as biologically important PAHs. Systematic studies with transition metals, ligand, base and solvent are employed to design a protocol for the transition-metal-catalyzed synthesis of nucleoside adducts. Identification of optimal conditions with respect to the variables leading to the synthesis of a library of compounds is the ultimate goal. We are also interested in the effect of ligand concentration on catalytic efficiency and product formation. The training of undergraduate students in a research lab is a very important goal of my research endeavor. The development of a special topic course on computational organic chemistry is envisaged. Such a course will significantly broaden the area of expertise of our chemistry majors in both experimental and theoretical techniques. Thus such broad expertise will make our chemistry seniors much better prepared for graduate school or to join the work force.
Sample Projects
Project 1
We are interested in understanding how, in a chimera of α- and β-peptide helices, various linkers constrain the spatial relationship between the segments. In this light the objectives were defined: (a) determine low-energy conformations for chimeric peptides linked by Gly or β-Ala (b) determine, for each system, whether conformations where the helices are roughly collinear (“unkinked” conformations) are particularly favored or disfavored (c) estimate how much enthalpic strain is required to “unkink” each chimeric peptide (d) estimate, as an entropic measure, the flexibility or rigidity of the low energy and “unkinked” conformations.
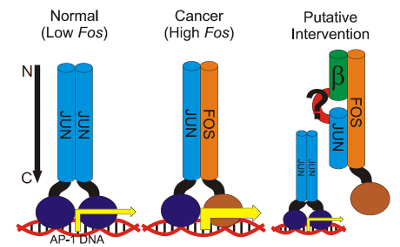
Model for the interaction of Fos and Jun dimers with DNA.
Project 2
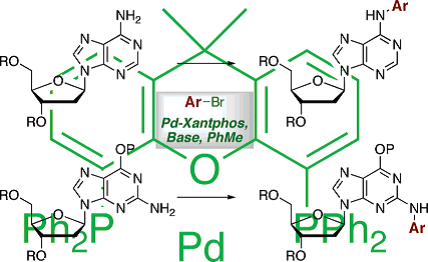
Direct arylation of the exocyclic amino groups of nucleosides represents a simple approach to N-aryl nucleoside derivatives. One limitation had been that only electron-deficient aryl bromides and triflates possessed adequate reactivity for efficient, direct N-arylation of nucleosides. We demonstrated that Pd-Xantphos catalytic systems lead to successful N-arylation of suitably protected 2’-deoxyadenosine and 2’-deoxyguanosine with a wide range of aryl bromides.
Project 3
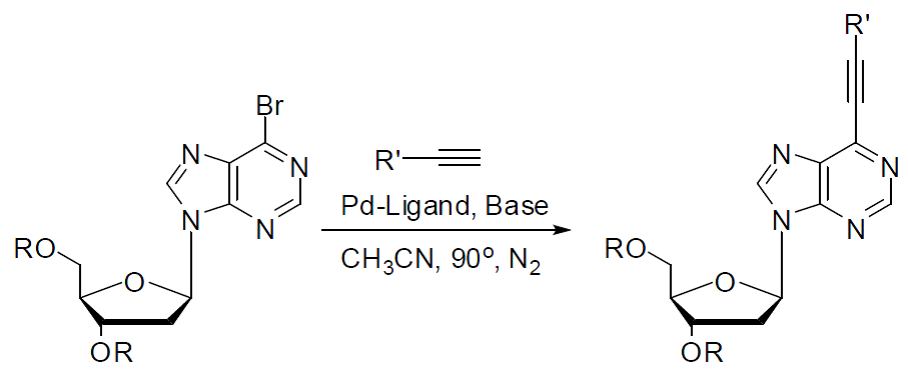
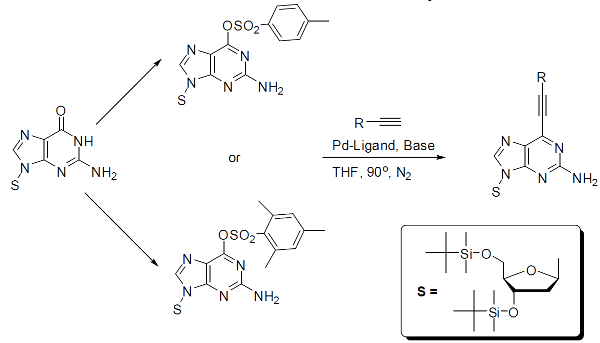
The Sonogashira cross-coupling reaction offers a convenient route to C(sp)–C(sp2) bond formation. Although the Sonogashira reaction has traditionally been carried out in the presence of Pd catalyst and a co-catalyst of Cu(I) salt, the use of Cu(I) salt is often not efficient because it leads to the formation of unwanted side-products. This has prompted interest in recent years in the development of Cu-free Sonogashira cross-coupling reaction conditions. In addition, the development of Cu-free Sonogashira cross-coupling conditions for the alkynylation of nucleoside derivatives remains largely unexplored. Herein, we demonstrate that Cu- and amine-free Sonogashira-type cross-coupling lead to successful alkynylation of aryl bromides and heteroaryl bromides. For the first time, we have extended this method for the alkynylation of protected 2’-deoxyadenosine at the C-6 position.
Project 4
The Sonogashira cross-coupling reaction provides a convenient route to C(sp)-C(sp2) bond formation. Traditionally, the Sonogashira reaction is carried out with a Pd catalyst, a Cu(I) salt co-catalyst, and an aryl or vinyl halide as a substrate. The Cu(I) salt co-catalyst is often not efficient because it promotes the formation of unwanted side products. Cu-free processes are therefore generally preferred for the Sonogashira reaction. Aryl sulfonates have been proven as an acceptable substrate for the Sonogashira reaction in place of aryl and vinyl halides. Shown here is the utilization of this means as a cleaner and more efficient alternative to halogenation in the modification of nucleosides. We have, therefore, shown that the sulfonation of protected 2’-deoxyguanosine provides a route to alkynylation at the C-6 position.
Project 5
β-Peptides are synthetic oligomers that mimic the secondary structures and folding arrangement of natural proteins. Fos and Jun are natural proteins that form heterodimers containing a leucine zipper region important for binding DNA and modulating transcription. We have performed docking simulations to model how b-peptides interact with each other and with the leucine zipper region of Fos or Jun. Brownian dynamics simulations of different β-peptides consisting of trans-2-aminocyclopentanecarboxylic acid (ACPC) or trans-2-aminocyclohexanecarboxylic acid (ACHC) provided starting snapshots of helix-helix complexes that were energy minimized to produce tightly packed dimers. The β-peptides of (R,R)-trans-ACPC bound the most intimately. For the most intimate, the two helices in the dimer ran antiparallel to each other. Simulations of β-peptides with Fos or Jun tested the ability of the β-peptide to interact with the leucine zipper regions. All β-peptides studied bound more favorably to Jun than to Fos with the electrostatic contribution to the potential energy making the difference.
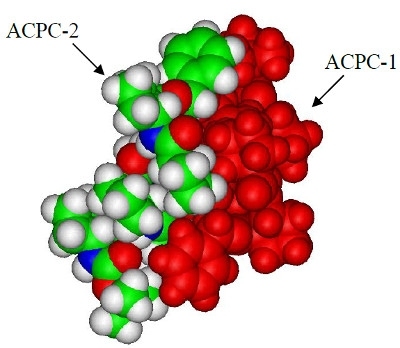
Most Intimate Dimer of (R,R)-trans-ACPC Hexamers. One hexamer (ACPC-1) is found on the right. The other hexamer (ACPC-2) is positioned on the left. The interaction energy is -37.3 kcal/mol and is dominated by the van der Waals interaction. Note how the benzyl group packs against the t-butyl group at each end of the dimer creating preferentialadocking.
Project 6
Much interest has been vested into the synthesis of the aryl ether linkage, which is commonly found in a large number of biologically active molecules. The traditional Ullmann coupling method for ether synthesis and palladium-catalyzed ether synthesis have limited applications. An efficient catalytic system has been developed for the synthesis of aryl ethers from various substituted phenol derivatives and aryl halides. The effect of different ligands, base, and solvent on the coupling of substituted phenol derivatives and aryl halides was investigated, as well as the effect of the amount of CuI and ligand.
[1475264391].png)
Mass spectrum of p-methoxy-diphenyl ether in dichloromethane
Project 7
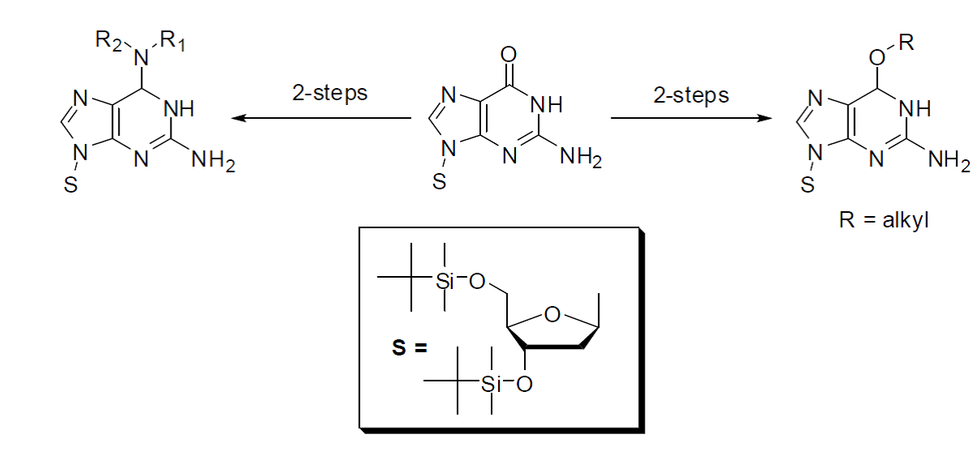
We have been able to convert the 3’,5’-bis-O-TBDMS derivative of 2’-deoxyguanosine to its O6-alkyl and O6-aryl ethers as well as to N6-substituted diaminopurine nucleosides in two simple steps. We also developed a novel, nonaqueous, one-step O6-desulfonylation method that leads to deprotection of the carbonyl moiety of 2’-deoxyguanosine.
Project 8
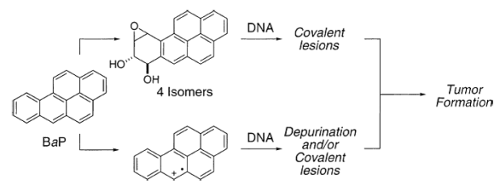
Single-electron oxidation of the carcinogenic hydrocarbon benzo[a]pyrene (BaP) is thought to result in a radical cation intermediate and this species has been proposed to cause alkylation at the nitrogens of the purine nucleobases. Although several different nucleoside adducts have been isolated as arising from this mode of metabolic activation, there are no selective, total syntheses of the stable exocyclic amino group adducts formed by the single-electron oxidation of any hydrocarbon with the purine 2’-deoxynucleosides to date. We reported the synthesis of the model adducts N6-(1-pyrenyl)-2’-deoxyadenosine and N2-(1-pyrenyl)-2’-deoxyguanosine as well as the first synthesis of the carcinogen-linked nucleoside derivatives N6-(6-benzo[a]pyrenyl)-2’-deoxyadenosine and N2-(6-benzo[a]pyrenyl)-2’-deoxyguanosine via a palladium-mediated C-N bond formation. Two different coupling strategies were attempted: coupling of an aryl bromide with a suitably protected nucleoside and the coupling of an arylamine with a suitable halonucleoside. The former had somewhat limited applicability in that only N6-(1-pyrenyl)-2’-deoxyadenosine was prepared by this method; on the other hand, the latter was more general. However, there are noteworthy differences in the amination reactions at the C-6 and C-2 positions. Reactions at the C-6 resulted in the competing formation of a 1:2 amine-nucleoside adduct in addition to the desired monoaryl nucleoside. Such a dimer formation was not observed at the C-2. The C-2 adducts, however, displayed an interesting conformational behavior.
Project 9
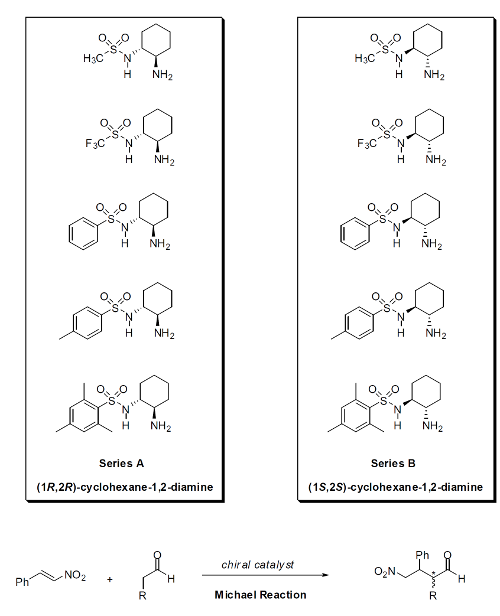
The synthesis of chiral sulfonamide organocatalysts of two series, series A and B, will be accomplished. These chiral organocatalysts will be examined in the conjugate addition (Michael addition) of aldehydes and ketones to nitroalkenes. This conjugate addition can be applied for the synthesis of γ-amino acids, which have been shown to be interesting foldamer building blocks.